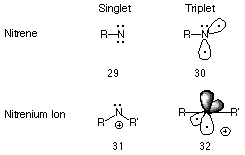
Isoelectronic with methylene, the nitrenium ion is an oft hypothesised reactive intermediate of many biological and laboratory processes. Just as nitrenes are the nitrogen analogues of carbenes, nitrenium ions are the analogues of carbocations or carbenium ions.92
Like nitrenes, these divalent positively charged species can exist in either the singlet or triplet state (Figure 1-7) and several papers have reported the semi-empirical93-95 and ab initio96-99 energy levels for the two states.100-104 MNDO studies of the parent alkylnitrenium ion NH2+ and alkyl-, dialkyl-, and alkylacylnitrenium ions (Ford and Scribner94) predicted ground-state triplets, whereas a singlet state has been proposed for aryl- and N-acylarylnitrenium ions.105
Recently, NH2+ has been the subject of extensive experimental and theoretical examination.105 High level ab initio calculations indicate that NH2+, like CH2+, has a triplet ground state and an ostensibly linear structure. The singlet-triplet separation So-T1 has been calculated by various semi-empirical and ab initio levels to be in the region of 30 kcal.mol-1.93,105
The chemistry of nitrenium ions has been reviewed and described a number of times since the early 1960's by Abramovitch,106,107 Lansbury,108 Gassman109-119 and McClelland.120 Recently Rudchenko121 has reviewed the synthesis, reactions and properties of ONO systems which also encompasses some of the chemistry of nitrenium ions.
Widespread interest in chemistry of these divalent positively charged reactive intermediates was sparked by the research of Gassman and co-workers.118,119 The basis for this work was to establish the migratory aptitudes of alkyl groups towards electron deficient nitrogen centres. It is known that alkyl groups tend to migrate to cationic but not to radical centres, and alkyl to nitrogen migration should be indicative of the presence of an electron deficient nitrogen. Edwards and co-workers122 had suggested that nitrenium ions could be generated from aliphatic chloramines using silver ions in polar solvents but he subsequently concluded that the reactions were homolytic at room temperature. Gassman and Fox on the other hand found that the methanolic solvolysis of 4,7,7-trimethyl-2-chloro-2-azabicyclo[2.2.1]heptane 33 in the presence of silver perchlorate afforded a mixture of products with the major component forming from alkyl to nitrogen rearrangement. In this reaction the methanolic solvolysis of 33 under reflux afforded 34 in 59% yield and 35 in 20% yield (Scheme 1-5).
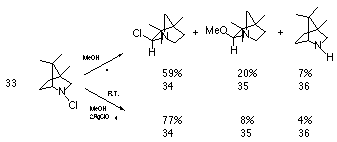
In the presence of silver perchlorate the reaction rate increased 2000 times and resulted in a different product distribution with a substantial increase in the yield of 34 at the expense of both 35 and the de-chlorinated product 36. Gassman et al concluded from these and similar reactions that, while the formation of a discrete nitrenium ion could not be positively proven, analysis of the reaction products implied the formation of a transitory electron deficient nitrogen.
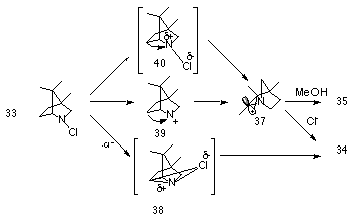
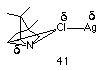
Gassman proposed two reaction pathways involving either a concerted rearrangement or a very tight ion pair 38 as illustrated in Scheme 1-6. Formation of 37 could occur via a concerted 1,2 alkyl to nitrogen shift with concomitant loss of chlorine 40 or alternatively from the discrete nitrenium ion 39 which is generated after heterolytic loss of chlorine from 33. The carbenium ion 37 would rapidly react with solvent or with chloride. The increase in the rate of solvolysis is clearly due to complex formation between the Lewis acid silver ion and chlorine, 41 which weakens the N-Cl bond and facilitates the migration.
The formation of a higher yield of chlorinated product as opposed to solvent trapped product is most probably due to a concerted reaction or a very tight ion-pair. While not predicted on the basis of nitrenium ion formation, a small yield of the reduced species 36 most probably arises from singlet to triplet nitrenium ion inter-system crossing followed by hydrogen abstraction.
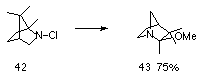
Other examples of silver-assisted 1,2-alkyl shift reactions have been reported, including the conversion of N-chloro-2-azabicyclo[2.2.1]heptane to exo-6-chloro-1-azabicyclo[2.2.1]heptane in 75% yield119,123 and the transformation of 42 to 43 (Scheme 1-7).
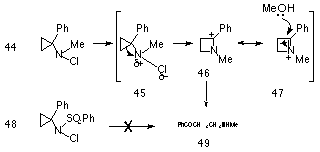
Deyrup et al124 found that electronic factors that manipulate the development of positive charge on the central nitrogen have a marked influence on the reactivity of the compound. While Gassman125 had observed the facile reactivity of N-chloro-N-methyl-1-phenylcyclopropylamine 44, Deyrup found the N-chloro-N-phenylsulphonyl derivative 48 to be rather inert to silver assisted solvolysis (Scheme 1-8). Clearly the presence of the strongly electronegative sulphonyl group hinders formation of the development of the positive charge on the adjacent nitrogen and retards the reaction process.
The solvolysis of N-chloroamines proved to be fertile ground for investigating 1,2-alkyl shifts and lead to a number of publications proposing the intermediacy of nitrenes or nitrenium ions. The reactions involving ring contractions126 and expansions127 were reviewed by Kovacic in 1970.128
Other sources of alkylnitrenium ions have been reported.
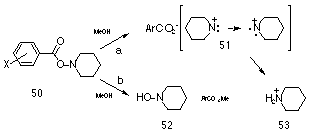
Gassman and Hartman117 solvolysed a series of O-benzoates of N-piperidine 50 in methanol and reported two reaction pathways: Scheme 1-9a in which a singlet piperidinyl nitrenium ion 51 forms which subsequently reverts to the triplet state which abstracts hydrogen; and Scheme 1-9b which provided the hydroxylamine 52 after methanolysis of the ester. Electron-withdrawing substituents would be expected to increase the polarisation of the carbonyl carbon facilitating nucleophilic attack by solvent, and indeed electron-withdrawing groups favoured pathway b over a. The linear free energy results for this series gave a sp correlation with r= + 0.68 (r = 0.995). The positive value for the slope is consistent with a transition state in which a positive charge is developing at nitrogen and the magnitude of r is half that expected in comparison to the ionic rearrangement of perbenzoates, which is similarly consistent with the ionic bond cleavage b- to the ring.
Gassman et al110,111 and Hobson and Riddell129 quickly realised the potential of nitrenium ions in the synthesis of heterocycles through intramolecular reactions. Subsequently the authors showed that N-chloroamines could be solvolysed to form azabicyclo compounds through a transannular cycloaddition with the help of silver ions (Scheme 1-10).
The addition of nitrenium ions to cyclic olefins, the so called pi-route to heterocycles, has proven controversial in that several authors believe the process is purely free radical in nature.123 While Gassman et al reported a series of synthetically useful, silver catalysed heterocyclic cycloadditions,110,111,114,130 Hobson et al objected to the participation of a discrete nitrenium ion in these processes since no reaction was evident when 57 was treated with silver perchlorate. However when AIBN was added, the chloramine reacted slowly to give the appropriate transannular cycloaddition129 (Scheme 1-11).
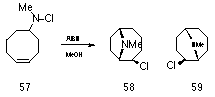
Hobson was prompted to remark that for chloroamines there appeared to be a "considerable predisposition towards homolysis, even in the reactions catalysed by silver salts". Further evidence has been presented by Edwards122 et al against the intermediacy of alkylnitrenium ions when, after a number of studies, they concluded that chloroamines do not form nitrenium ions near room temperature even in the presence of available double bonds and silver ions and that the reactions are homolytic.
The investigation into the properties of aryl stabilised nitrenium ions has been intensive in the last decade due in part to their postulated intermediacy in biological processes that lead to mutagenesis. Arylnitrenium ions 60 are characterised by delocalisation of the positive charge on nitrogen into the aromatic ring (Scheme 1-12).
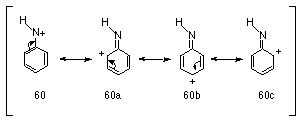
Arylnitrenium ions were first implicated in the reactions of aryl azides with nucleophiles in acid solutions131,132 (Scheme 1-13).
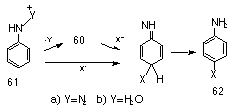
While the reaction for 61a was said to be either a concerted or two-step reaction, the driving force for the process is clearly the loss of nitrogen. Kinetic studies133 on the acid-catalysed solvolysis of arylhydroxylamines 61b, indicated formation of the nitrenium ion via an SN1 process. These arylnitrenium ions are extremely long lived in water,134-140 especially when compared to carbenium ions.120 McClelland recently determined the lifetime of nitrenium ions formed in the Bamberger rearrangement of N-arylhydroxylamines to be 125-250 ps.136 With para-alkoxy substituents, lifetimes as long as 1 ms have been observed.141
In the early 1970's, Gassman et al studied the decomposition of N-chloro-N-alkylanilines 62 and inferred that arylnitrenium ions 63 where intermediates in the process.142
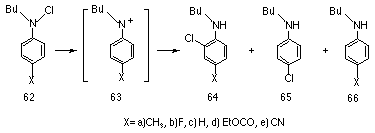
The solvolysis of 62a-e in buffered ethanolic solutions gave a mixture of products, with the major one in all reactions being the o-chloroaniline derivative (64, 65-85%) (Scheme 1-14).
A Hammett plot revealed an excellent relationship between log kx and s+p (r = -6.4, r=0.996) which indicated the formation of a positively charged intermediate species. With increasing electron-withdrawing para substituents, the nitrogen would be expected to bear more of the positive charge and subsequently the yield of the reduction product 66 increased from 1% for 62a to 29% from 62e. The higher yield of the reduction product (which derives from either direct hydride transfer from the solvent or through the increase in intersystem crossing to the triplet form, leading to hydrogen abstraction and deprotonation) is a consequence of the increased nitrenium ion character.
Formation of the nitrenium ion is facilitated by delocalisation of the positive charge onto the aromatic ring and this resonance interaction is promoted by electron-donating substituents. Supporting evidence for delocalisation of charge from the nitrenium ion onto the ring was found with the isolation of 10% of 67 from the ethanolic solvolysis of 62a and 32% of 68 from the methanolysis of the same compound.
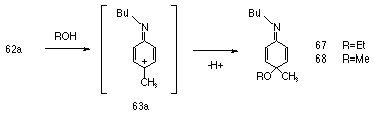
There is evidence to suggest that an arylnitrenium ion is best viewed as a cyclohexadienyl cation bearing an imine substituent, such as 63a in Scheme 1-15, and in support of this, the following observations have been made by McClelland:120 substituents on the nitrogen have little influence over the lmax of the cation;134,143 substituents para to nitrogen have a marked influence120 over reactivity; p-methoxyphenylnitrenium ion was observed as an O-methylated-2,5-cyclohexadienone like structure from irradiation of the precursor azide in water.120 Recently Novak has presented preliminary ab initio calculations which suggest that resonance structure 60b is considerably more important than for the corresponding carbenium analogue.144
A large number of similar studies dealt with the silver ion promoted nucleophilic aromatic substitution of N-chloroamines,130,142,145 such as that illustrated in Scheme 1-16.
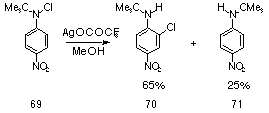
They indicated that the yield of reduction and o-chlorinated products increased and that of solvent trapped product decreased markedly as electron-withdrawing substituents were introduced. Formation of a delocalised arylnitrenium ion was facilitated by formation of a tight silver ion pair, which undergoes attack by solvent, generally at the para position. Formation of the o-chlorinated product follows from this closely constrained, tight ion pair. With increasing electron-withdrawing substituents on the ring, the stability of the arylnitrenium ion decreases resulting in lower charge on the ring and a greater positive charge on the nitrogen. They proposed that a more positive nitrogen would lead to a tighter ion pair with silver, thereby facilitating o-chlorination at the expense of nucleophilic attack from solvent.
Intramolecular trapping of nitrenium ions offers great synthetic potential.123,146-150
Model proximate carcinogens
Carcinogenic aromatic amines and amides are believed to be transformed in vivo to an ultimate carcinogenic metabolite (see also Chapter 4).61,64,65 Some important polycyclic aromatic amines and amides including 2-aminofluorene and N-acetyl-2-aminofluorene 18 are known to be metabolised into potent carcinogens in laboratory animals.61,64
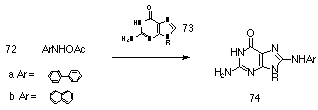
O-acetyl-N-arylhydroxylamines (such as 72a and 72b) are putative metabolites of polycyclic aromatic amines and react at the 2'-deoxyguanosine base to generate C-8 adducts in low yields151,152 (Scheme 1-17). In mixed organic solvents, 72 react in a similar fashion with guanosine or 2'-deoxyguanosine to generate 74 in a process assumed to involve a DN + AN (SN1) mechanism in which there is highly selective trapping of nitrenium ion generated by rate-limiting N-O bond heterolysis.153 Novak proposed that the carcinogenic potential of 72a and 72b in aqueous solution is due to this selectivity for C-8 of 2'-deoxyguanosine.
Extensive studies of the carcinogenic metabolites, including sulfuric and carboxylic acid esters, of N-acetylaminofluorene have recently indicated that the nitrenium ion is extremely long lived in water (lifetimes of approximately 30 ms). This has been attributed to severe disruption to aromatic character if attack by solvent occurs at the aromatic ring. Deoxyguanosine however reacts at diffusion controlled rates with N-acetyl-N-fluorenylnitrenium ion to give the C-8 adduct 76. This unusual reactivity pattern is currently unexplained.
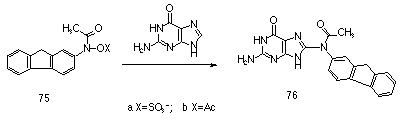
Other sulfuric acid esters of N-phenyl-N-hydroxyamides64,88 have been investigated extensively by Novak et al.88,90,154,155 In aqueous solution these compounds undergo N-O bond cleavage to yield products characteristic of processes involving nitrenium ion-sulphate ion pairs.82,154,156-161
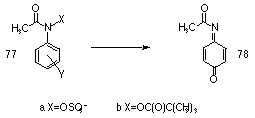
Several labile intermediates, including benzoquinone imines 78, are detected during hydrolysis of 77 and these are considered to play an important role in the in vivo chemistry for these sulfuric acid esters.89,162 Novak proposed that the tight ion pairs undergo internal return to yield rearrangement products and solvent separated ion pairs which are attacked by external nucleophiles or reducing agents.90,154,155
A series of ring substituted N-sulfonoxyacetanilides were synthesised to serve as models for the carcinogenic metabolites of polynuclear aromatic amides by Novak et al.84-86,88-90,154,155,162-166 Kinetic and product studies in aqueous solution indicated that the hydrolysis reactions display moderate pH dependence91 and that nitrenium ions derived from hydrolysis of N-(sulfonatooxy)-N-acetyl-2-aminofluorene 75 and N-(sulfonatooxy)-N-acetyl-2-aminobiphenyl can also be trapped by the glutathione anion.167
In parallel with the work on N-(sulfonatooxy) derivatives, Novak investigated potential models for the acylated metabolites of the polycyclic amines using N-aryl-O-pivavloylhydroxylamines 77b. These compounds undergo SN1 alcoholysis to give nitrenium ions in a similar fashion to the related N-(sulfonatooxy) derivatives. However this process can be influenced by experimental conditions which can be adjusted to suppress nitrenium ion formation and enhance the bimolecular process. For instance, in the reaction between N-aryl-O-pivavloylhydroxylamines 79 and N-N-dimethylaniline85,86 in methanol, second-order kinetics were observed and the kinetic products were generated from the nucleophilic attack at the nitrogen of the hydroxylamine derivative (Scheme 1-20).
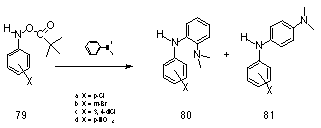
79a-d were found to react by an SN2 mechanism with N,N-dimethylamine in MeOH to furnish the adducts 80 and 81 in high yield (Scheme 1-20). This was the first example of nucleophilic substitution with bimolecular kinetics in ester derivatives of N-arylhydroxylamines.86,168 Subsequently, bimolecular kinetics for the reaction of N-methylaniline with N-(cyanophenyl)-O-(diphenylphosphinoyl)hydroxylamine were also reported by Boche.83
In summary, carboxylic or sulfuric esters of N-arylhydroxylamines and N-arylhydroxamic acids have been implicated as the ultimate carcinogens derived from the metabolic activation of aromatic amines and amides and these compounds generate nitrenium ions by rate-limiting N-O bond cleavage during solvolysis. These nitrenium ions have long lifetimes in solution and in certain cases are efficiently trapped by deoxyguanosine under experimental conditions designed to mimic physiological processes. While impressive advances have been made in understanding the reaction mechanisms for these highly reactive metabolites, unequivocal evidence for SN1 and SN2 reactivity in vivo has not yet been presented.
Glover has proposed that nitrenium ions (Figure 1-7) should be stabilised by neighbouring nitrogen, phosphorous, oxygen and sulfur atoms as the lone pairs can overlap strongly with the vacant 2p orbital on nitrogen. Extensive MNDO and ab initio molecular orbital calculations by Scott105 and Glover95 had indicated a strong resonance interaction between the nitrogen and these heteroatoms (Figure 1-9).
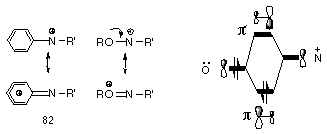
They predicted that the strong double bond character would lower the energy of the nitrenium ion and impart some degree of stabilisation to an otherwise very reactive intermediate.
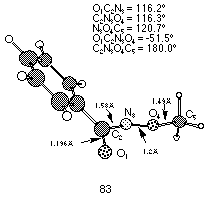
AM1 calculations on N-benzoyl-N-methoxynitrenium ion 83 indicated that the positive charge, while nominally situated on the nitrogen, is effectively spread over the methoxy moiety (Table 1-2). AM1 gas-phase atomic charges indicate that 0.48 of a positive charge is delocalised over the methoxy side-chain, primarily due to the strong overlap of N2pz and O2pz orbitals, resulting in substantial pi-bond character calculated to be as high as 0.9.95
|
Group |
|
|
N |
|
|
CO |
|
|
C6H6 |
|
|
OMe |
|
They were able to generate N-alkoxy-N-benzoylnitrenium ions 85 by treatment of the alkyl N-chlorobenzohydroxamate species 84 with Lewis acids such as silver salts (Scheme 1-21) and used them as electrophiles in intramolecular cyclisation reactions.
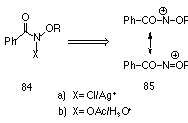
Glover and Scott also investigated the consequences of resonance stabilisation for the mode of cyclisation onto aromatic rings.95,105,169,170 The intrinsic mesomeric stabilisation of an electron deficient nitrogen by the lone pair of a neighbouring heteroatom can be invoked to explain the different modes of reaction.
Cyclisation of alkoxy-stabilised nitrenium ions onto aromatic rings has been used in the preparation of heterocycles and in particular, the synthesis of novel benzoxazines and benzoxazepines.169-171
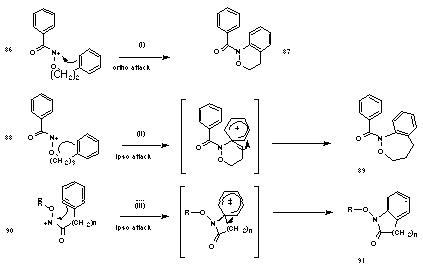
Glover found that N-acyl-N-(3-phenylpropyloxy)nitrenium ions 88 cyclise to 1,3,4,5-tetrahydro-2,1-benzoxazepines 89 through ipso attack followed by 1,2 carbon migration (Scheme 1-22(ii))169,170 whereas N-acyl-N-(2-phenylethoxy)nitrenium ions 86 cyclise directly at the ortho position to give 3,4-dihydro-1H-2,1-benzoxazines 87 (Scheme 1-22(i)), rather than through ipso cyclisation. The difference lies in the ability of the six-membered, as opposed to the five-membered ring, to accommodate the endocyclic +N-O p-bond character. N-alkoxylactam 91 formation involves ipso attack even when a 5-membered transition state is needed (Scheme 1-22(iii)). The exocylic NO pi-bond, is no impediment to such transition state formation.
Theoretical calculations at the MNDO level also revealed that the stabilisation imparted by the lone pair of the oxygen parallels that imparted by N-aryl substituents. The N=Cipso pi-bond orders for 82 and for N=O in 85 are both calculated to be 0.90. Furthermore, Glover proposed172 that an N-alkoxy stabilised nitrenium ion precursor such as alkyl N-acetoxybenzohydroxamates should also display mutagenic behaviour by analogy with N-acetoxy-N-arylamides (see also Section 2.2.1). The similar stability and ease of formation of N-alkoxy and N-arylnitrenium ions suggested that such compounds might behave as electrophiles towards DNA.
Accordingly, butyl N-acetoxybenzohydroxamate (84b,
R=Bu) was synthesised and found to be mutagenic. Preliminary
experiments under acidic conditions indicated that alkoxy stabilised
nitrenium ions were generated under solvolysis (Scheme 1-21), as
predicted by semi-empirical calculations.
The goals of this project are: