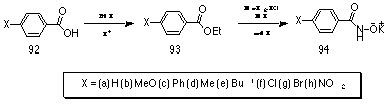
|
|
|
The aim of this thesis is an investigation of the reactivity of alkyl N-acyloxybenzohydroxamates. This new class of mutagens were first synthesised by Glover172 by acetoxylation of alkyl benzohydroxamates with silver acetate, but this procedure was subsequently found to be unsuccessful in a number of cases. Thus a versatile synthesis of a wide range of mutagens was developed, based on work by and Cooley.173 The versatility of this new synthesis arises from the ease of variation in the alkyl and acyl groups, with the potential for a wide range of "designer mutagens" to be developed.
Potassium benzohydroxamate salts 94 were obtained through treatment of the appropriate ethyl benzoate esters 93 with hydroxylamine hydrochloride under basic conditions in methanol. The salts precipitated in moderate to good yields over time with refrigeration.
Ethyl esters 93 were obtained by classical esterification of the appropriate benzoic acids 92 with excess ethanol under acidic conditions and were purified by distillation and characterised by b.p., IR, 1H and 13C NMR spectroscopy.
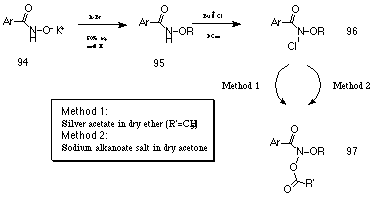
Alkyl benzohydroxamates 95 were synthesised in good yield by alkylation of the potassium benzohydroxamate salt 94 with bromoalkane in refluxing 50% aqueous methanol (Scheme 2-2). Generally, alkyl bromides were either purchased or synthesised by bromination of the corresponding alcohol using hydrobromic acid.
Simple straight chain alkyl benzohydroxamates 95 were found to be light, clear oils which were unstable and degraded over several weeks to complex mixtures. As a result, benzohydroxamic esters were usually purified by flash column chromatography immediately prior to use. Several butyl para-substituted benzohydroxamic esters were obtained as low melting point solid compounds that could be satisfactorily recrystallised from chloroform and hexane.
All alkyl benzohydroxamates 95 were satisfactorily characterised by b.p. or m.p., IR, microanalysis, mass spectra and NMR spectroscopy.
Alkyl benzohydroxamates 95 were first converted to their N-chloro derivatives 96 using a 3M excess of t-butyl hypochlorite at room temperature in dichloromethane (Scheme 2-2). Reaction times varied from 2-12 hours but 1H NMR provided a convenient diagnostic test to determine the progress of the reaction. Chlorination of the alkyl benzohydroxamates 95 generally resulted in a downfield shift of approximately 0.15 ppm to the 1'-methylene protons of the alkyl moiety. Removal of solvent in vacuo provided the alkyl N-chlorobenzohydroxamates 96 as yellow oils that could not be purified by flash chromatography but were sufficiently clean to be used immediately without further purification. Two general methods were used for acetoxylation of the N-chlorobenzohydroxamic esters (Scheme 2-2).
Method One developed by Glover and co-workers172 involved treating the alkyl N-chlorobenzohydroxamates 96 with a 2 molar equivalent of the Lewis acid, silver acetate, in dry ether. The mechanism of acetoxylation probably proceeds via a silver catalysed heterolysis of the N-Cl bond to form a nitrenium ion that is trapped by the acetate anion. Where this method was successful, fair to good yields were obtained.
Method Two involved stirring 96 at room temperature with a 1.4 molar excess of anhydrous sodium alkanoate salt for 12-48 hours in anhydrous acetone. Solid NaCl crystallising above the surface of the dry acetone was an excellent indicator that the N-acylation was proceeding smoothly. The acylation reaction could not be allowed to stir indefinitely in dry acetone as the yield of alkyl N-acyloxybenzohydroxamate 97 reached a maximum and then slowly reduced. An excess of sodium acetate significantly greater than 1.4 molar generally reduced the yield of 97 and increased the amount of (principally benzoate ester) contaminants. The mechanism of acetoxylation for Method Two most probably involves an SN2 process.
The progress of N-acyloxylation was monitored by reverse-phase HPLC analysis or in the case of alkyl N-acetoxybenzohydroxamates, through 1H NMR spectroscopy by observing the formation of the resonance for the N-acetoxy peak at d2.05.
Filtration and removal of solvent in vacuo at room temperature provided the crude alkyl N-acyloxybenzohydroxamates which were purified by flash chromatography in fair to excellent yields. Potential instability and possible hazards precluded mass spectra and microanalytical analysis, and 97 were satisfactorily characterised from IR, 1H, gated decoupled and where needed JCH correlated 13C spectra at 298K. The infrared spectra for alkyl N-acetoxybenzohydroxamates displayed two strong characteristic carbonyl absorptions at approximately 1730 cm -1 and 1790 cm-1. The high amide carbonyl absorptions at 1730 cm-1 are indicative of a low amide isomerisation barrier. This has been interpreted to indicate a pyramidal nitrogen geometry and these compounds are considered to be members of the class of the so-called "anomeric amides". Anomeric effects are possible in such compounds and lead to significantly increased N-OR rotational barriers.174,176,177
The non-diastereotopic nature of the methylene protons NO-CH2Ph on the alkyl side chain which was evident from the single sharp resonance at ca. d 5.2 in the 1H NMR is however indicative of fast nitrogen inversion for this class of hydroxamic ester. Asymmetric non-bridgehead nitrogen compounds have been reported for systems with similar geminally substituted nitrogen174 however the pyramidal stability of ONO systems is greatly affected by the nature of the substituents and steric effects.121 The rate determining step in the topomerization of ONO systems is the inversion at nitrogen which proceeds via a planar transition state in which the lone pair occupies the Npz orbital. Electronegative ligands increase the s character of the nitrogen non-bonding orbital, which in turn increases the energy of the inversion transition state, disfavouring a fast inversion-rotation process. For both alkyl N-chlorobenzohydroxamates 96 and alkyl N-acetoxybenzohydroxamates 97 the combined electronegativity of the alkoxy and chloro or acetoxy functional groups were clearly insufficient to restrict nitrogen inversion, resulting in the sharp oxymethylene resonances. The inversion barriers in these amide systems121,175 are most probably lowered since in the planar transition state, conjugation is possible. These barriers were recently investigated by Rauk and Glover who found them to be in the region of 2-3 kcal.mol-1 and did not vary significantly over a range of anomeric amides.176
Alkyl N-acyloxybenzohydroxamates 97 are relatively stable compounds if stored under nitrogen and refrigerated. Under these conditions they decompose to alkyl benzoate esters and acetic acid over several weeks.