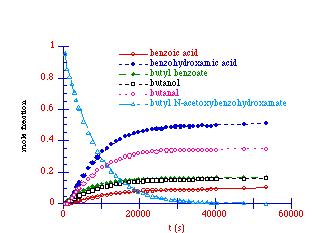
An analysis of the 1H NMR spectrum at the completion of a typical rate study run for the solvolysis of butyl N-acetoxybenzohydroxamate at 308K revealed the presence of a number of solvolysis products which were benzoic acid, benzohydroxamic acid, butyl benzoate, butanol and butanal. All products were readily identified in the 1H spectrum and their presence was confirmed by 13C spectroscopy by comparison with authentic samples under identical conditions (D3-acetonitrile/D2O)
Solvolysis products for butyl N-acetoxybenzohydroxamate at 308K
A solvolysis reaction for 100a was initiated at 308K and the disappearance of the mutagen and formation of the products was monitored over 15 hours and is shown in Figure 2-14.
Analysis of the formation of products indicated parallel formation in concert with the disappearance of starting material. For parallel product formation from unimolecular reactions, Scheme 2-8 pertains.
As the concentration of all products is zero at the start of the reaction, ie. [prod]o = 0, for all products (1, 2 and 3) the following holds:
A plot of the appearance of the solvolysis species versus ![]() for the solvolysis
of butyl N-acetoxybenzohydroxamate 100a is shown in
Figure 2-15. In this plot, the yield of each product has been
determined from the integral of the signal in the 1H NMR and has been subsequently normalised with
respect to the initial concentration of the N-acetoxy
compound. Hence, at t=0, the concentration of 100a (also
shown) is 1.0.
for the solvolysis
of butyl N-acetoxybenzohydroxamate 100a is shown in
Figure 2-15. In this plot, the yield of each product has been
determined from the integral of the signal in the 1H NMR and has been subsequently normalised with
respect to the initial concentration of the N-acetoxy
compound. Hence, at t=0, the concentration of 100a (also
shown) is 1.0.
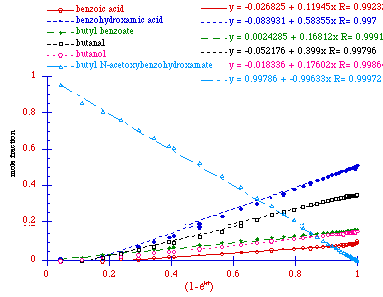
Figure 2-15 reveals that the data for all species simplify to
straight lines upon plotting the normalised concentrations
against![]() as the abscissa and the partial
rate constants for appearance of the products are obtained from the
individual slopes,
as the abscissa and the partial
rate constants for appearance of the products are obtained from the
individual slopes, ![]() . The acid
independent rate of solvolysis for butyl
N-acetoxybenzohydroxamate in this reaction was
(2.51±0.02) x 10-2 s-1. The sum of the relative rate constants for
formation of butyl benzoate, benzoic acid, and benzohydroxamic acid
is close to the overall rate constant, since the sum of ratios
(kprod:k) is almost 0.9.
. The acid
independent rate of solvolysis for butyl
N-acetoxybenzohydroxamate in this reaction was
(2.51±0.02) x 10-2 s-1. The sum of the relative rate constants for
formation of butyl benzoate, benzoic acid, and benzohydroxamic acid
is close to the overall rate constant, since the sum of ratios
(kprod:k) is almost 0.9.
Butanol and butanal must be formed in conjunction with formation
of benzohydroxamic acid and benzoic acid. Butyl benzoate is
not formed from butanol and benzoic acid, since the alcohol,
carboxylic acid and ester are formed in parallel reactions. This is
clearly seen in Figure 2-15 as the ratio of the yield of all three
remain constant, signified by the straight line relationship between
the concentrations of all three with respect to ![]() .
.
Further insight into the mechanism of decomposition was obtained from studies of the pH dependence of product formation.
Acid dependence of product distribution
Butyl N-acetoxybenzohydroxamate 100a was solvolysed at five acid strengths to determine the effect of pH on the percentage yields of the solvolysis products. The results are given in Table 2-3 and plotted in Figure 2-16.
|
|
|
| ||||
|
|
|
|
|
|
|
|
|
0.003343 |
0.004963 |
|
|
|
|
|
|
0.005028 |
0.007579 |
|
|
|
|
|
|
0.016650 |
0.019010 |
|
|
|
|
|
|
0.050820 |
0.065096 |
|
|
|
|
|
|
0.100890 |
0.122870 |
|
|
|
|
|
a Percentage yields were calculated by integration of appropriate resonances in the 1H NMR spectrum at the completion of the reaction and comparison with the integration of the acetoxy singlet for butyl N-acetoxybenzohydroxamate at the start of the solvolysis.
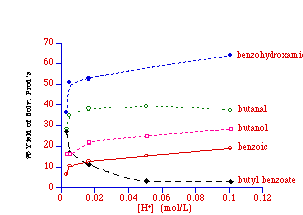
Increasing sulphuric acid strength resulted in higher yields of benzoic acid, benzohydroxamic acid and butanol. The yield of butanal also increases as pH decreases but appears to reach a maximum value and then decrease slightly. This apparent maximum yield is most probably an artefact of the error associated with obtaining an accurate integral of the methylene hydrogens (ca. d2.3) adjacent to the aldehyde group in the 1H NMR spectrum. At high acid concentrations there was noticeable broadening of this triplet signal. While the integral of the signal should not be affected by loss of resolution brought on by line broadening, there is an increase in the error associated with the integral and hence the yield. The formation of these products has clearly been promoted by a decrease in pH.
The yield of butyl benzoate on the other hand, clearly decreases as the acidity increases, indicating that formation of the ester is an acid-independent process and thus the formation of ester does not compete with the acid-catalysed processes at higher acid concentration.
Dependence of product distribution upon electronic effects
The product distribution at the end of suitable kinetic runs was analysed for the series butyl N-acetoxy-para-substituted benzohydroxamate (p-MeO, p-Me, p-H, p-Cl, p-Br, p-NO2) and the results are displayed in Table 2-4. While pH was not identical in each case, some important observations could be made.
|
X |
|
|
|
|
|
|
|
[H+]/10-3 molL-1 |
2.2 |
8.5 |
3.3 |
9.4 |
4.2 |
8.3 |
|
Butanol |
16 |
22 |
16 |
25 |
16 |
22 |
|
Benzoic acid |
4 |
4 |
6 |
17 |
a |
a |
|
Butanal |
34 |
31 |
28 |
21 |
16 |
0 |
|
benzohydroxamic acid |
19 |
21 |
36 |
32 |
a |
a |
|
Butyl benzoate |
9 |
11 |
27 |
29 |
42 |
42 |
a Could not be determined due to overlapping resonances.
Butanol is produced in similar, moderate quantities by all para-substituted compounds. Benzoic acid was found in low yields with electron-donating substituents and appears to be produced in greater quantities under the influence of electron-withdrawing substituents, although data for p-bromo and p-nitro substrates could not be obtained. Conversely, butanal is produced in fair yields with electron-donating substituents but the yield is reduced with electron-withdrawing substituents and, significantly, is zero with p-nitro. Benzohydroxamic acid is found in moderate yields across the series and in yields at least similar to those of butanal in the case where measurement was possible. The yield of butyl benzoate was markedly influenced by the para substituents and is formed preferentially where electron-withdrawing groups are present.
In aqueous acetonitrile, rate determining formation of nitrenium ion is most probably followed by a rapid solvolysis of the positively charged nitrenium ion intermediate. Subsequent loss of a hydrogen ion would provide the hitherto unknown intermediate, N-butoxybenzohydroxamic acid 108.
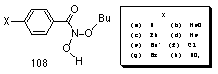
AM1 heats of formation for important intermediates along this pathway for the model methyl N-acetoxybenzohydroxamate 109 are depicted in Scheme 2-9.
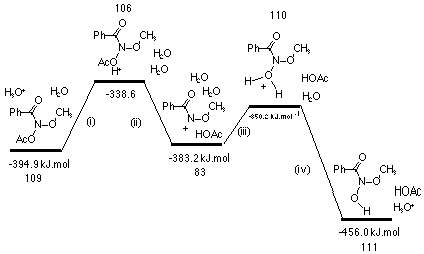
AM1 calculations on methyl N-acetoxybenzohydroxamate 109 indicated that acid-catalysed nitrenium ion formation was possible. Rate determining formation of the activated complex occurs upon reversible protonation of the N-acetoxy carbonyl carbon 106 which subsequently loses acetic acid to form the reactive intermediate nitrenium ion 83. According to these semi-empirical calculations, hydrolysis of the nitrenium ion appears to be a favourable process. These results indicate that the heterolysis is essentially a thermoneutral process and that overall, formation of N-methoxybenzohydroxamic acid 111 is strongly exothermic (Scheme 2-9).
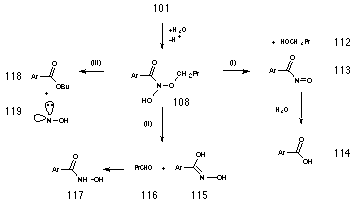
N-butoxybenzohydroxamic acid 108 could not be isolated from the reaction mixture, however unequivocal evidence for it's formation was provided by 18O isotope studies described in Section 2.2.10. Nor could it be detected in situ during the reaction rate studies which were monitored continually by NMR. The decomposition pathways proposed for 108 are illustrated in Scheme 2-10 and, together, satisfactorily account for all the products as well as the effects of acid concentration and para substituents.
Acid catalysis could lead to the formation of butanol 112 and nitrosocarbonylbenzene 113 in a similar process to acid-catalysed decomposition of hemiacetals194 (Scheme 2-10i). Nitrosocarbonylbenzene could then be the source of benzoic acid 114 through reaction with water. Alternatively, protonation at the benzoyl carbonyl could lead to loss of butanal 116 and oxime 115 which would rearrange to the more stable tautomer, benzohydroxamic acid 117 (Scheme 2-10ii). The conjugate anion of the alkoxy benzohydroxamic acid intermediate could also result in the formation of ester through a novel internal rearrangement releasing hydroxynitrene as a reactive by-product (Scheme 2-10iii). All three possible mechanisms are addressed below.
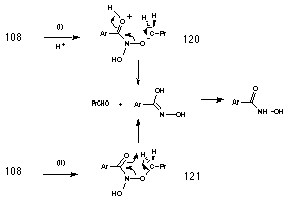
Butanal and benzohydroxamic acid appear to be generated together in an acid-catalysed elimination reaction (Table 2-3 and Figure 2-16). The yields of products (estimated by 1H NMR) from the solvolysis of butyl N-acetoxybenzohydroxamate 100a at different acid concentrations are illustrated in Figure 2-16. There is an increase in both aldehyde and hydroxamic ester formation with increasing acid strength in accord with acid-catalysis. Two possible mechanisms for the formation of butanal and benzohydroxamic acid are shown in Scheme 2-11. Scheme 2-11(i) is an acid catalysed reaction in which the protonated intermediate N-butoxybenzohydroxamic acid 120 rearranges to give the required products. As an acid catalysed process, the reaction will be favoured with a decrease in pH. Scheme 2-11(ii) is an acid independent mechanism where rearrangement occurs via the formation of a six-membered transition structure 120 allowing internal transfer of a proton between butoxy and carbonyl moieties. An increase in acid concentration would favour other acid catalysed solvolysis processes over this mechanism and thus the yield of butanal and benzohydroxamic acid would have decreased.
The yield of both solvolysis products increases as the concentration of hydronium ion increases, indicating that Scheme 2-11(i) holds over both Scheme 2-11(ii) and direct elimination from the free nitrenium ion.
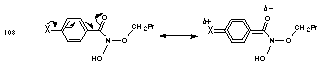
The basicity of the benzoyl oxygen will be influenced through mesomeric and inductive interactions from substituents on the benzoyl ring. Electron-donating substituents para to the carbonyl group will reduce the partial positive charge associated with the normal carbonyl polarisation and increase the basicity of the carbonyl oxygen (Figure 2-17). Positive mesomeric groups will also increase the electron density on the carbonyl oxygen through direct resonance interactions across the ring and into the carbonyl moiety.
From Table 2-4 the yield of butanal is greatest (34%) with a para-methoxy substituent 100b. The data for benzohydroxamic acid is incomplete due to the overlap of benzoic and benzohydroxamic acid resonances in the 1H NMR spectrum and errors in 1H NMR integrations were appreciable. Nevertheless, butanal and hydroxamic acid are formed in relatively similar yields. The yield of butanal progressively decreased as the electron-donating ability of the para substituent diminished. This is most evident when p-nitro 100h was present on the ring in which case the yield of butanal dropped to zero. Clearly, the ring substituent influences the basicity of the carbonyl oxygen which controls the mechanism leading to formation of butanal and benzohydroxamic acid.
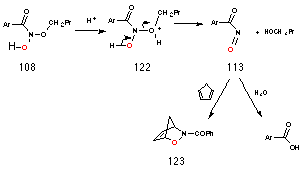
The source of butanol is most likely acid-catalysed decomposition of N-butoxybenzohydroxamic acid 108 to the nitrosocarbonylbenzene 113 intermediate which, like acyl chlorides, reacts with water195 to give the benzoic acid (Scheme 2-12). The yields of benzoic acid and alcohol for butyl N-acetoxybenzohydroxamate 100a also improve with increasing acid concentration (Table 2-3). Protonation of the butoxy oxygen 122 would be expected occur at higher acid concentration thus promoting formation of butanol and nitrosocarbonylbenzene.
The formation of nitrosocarbonylbenzene 113 as a key intermediate in the solvolysis reaction was confirmed by selectively trapping the nitrosocarbonylbenzene intermediate, as a Diels-Alder adduct, from the solvolysis mixture.
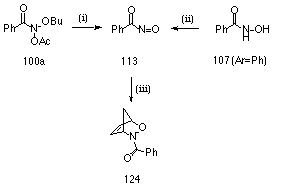
Nitrosocarbonylbenzenes are transient intermediates formed by oxidation of benzohydroxamic acids and are excellent dienophiles196-205 Addition of cyclopentadiene to the reaction mixture for the acid-catalysed solvolysis of butyl N-acetoxybenzohydroxamate 100a in 25% aqueous acetonitrile led to the formation, albeit in low yield, of N-benzoyl-2,3-oxazabicyclo[2.2.1]hept-5-ene 124 (Scheme 2-13i). This was detected by both NMR and HPLC and was identical (NMR and retention time) to authentic material prepared by oxidation of benzohydroxamic acid with N-bromosuccinimide in dichloromethane in the presence of cyclopentadiene (Scheme 2-13ii).197 The authentic material was subjected to a number of NMR studies (1H, 13C, Gated decoupled, COSY and NOESY) to confirm its structure. The adduct was isolated by preparative HPLC (1.4%) and displayed an extremely weak (M+2)+ ion in its mass spectrum. A similar trapping reaction in 25% (10% 18O enriched) aqueous acetonitrile afforded the adduct which displayed an increased intensity for the (M+2)+ ion. Here the (M+2)+:M+ ratio was 0.148 in its mass spectrum. While the error in this ratio is quite large since the adduct displayed a very weak molecular ion under electron impact (only 2.7% of base peak), best estimates of the corresponding ratio at natural abundance in the pure standard adduct were 0.023 (based on an M+ of 4.3%) and 0.024 (based on an M+ of 4.2%) in two independent measurements, while the theoretical value is only 0.0126.
The extent to which 18O is incorporated provides clear evidence for both the trapping of the nitrenium ion intermediate by solvent water molecules to give N-butoxyhydroxamic acid 108 and subsequent hemiacetal-like acid-catalysed decomposition to the nitrosocarbonylbenzene 113. While the yields of benzoic acid 114 are erratic, those for butanol are relatively similar across the para-substituted series (Table 2-4). Substituents would not be expected to influence the protonation step involved in this process. Poor yields of benzoic acid might be ascribed to both difficulties in NMR measurement across the series as well as other, hitherto undetected reactions of the nitrosocarbonylbenzene intermediate.
Esters 118 are not formed from alcohol and benzoic acids, since alcohols and esters are formed in parallel processes (Figure 2-15).180 In addition, Figure 2-16 indicates that there is a large reduction in ester formation with increasing acid concentration in accordance with a non acid-catalysed process in competition with acid-catalysed pathways. From these observations it appears plausible that esters are formed in a concerted, intramolecular rearrangement resulting in the formation of hydroxynitrene 119 (Scheme 2-14).
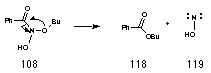
Several precedents for this type of process have been discovered in these laboratories and the process appears to be a general one for N,N-bishetereoatomsubstituted amides 177.206 Called the HERON reaction, it is favourable in most RCONXY systems where the lone pair on Y can lead to an anomeric weakening of the N-X bond.207 Such interactions are best with high energy lone-pairs (nY) and low energy s*N-X orbitals and the HERON reaction was first discovered when alkyl N-acetoxybenzohydroxamates 97 were reacted with N-methylaniline.208 The product of SN2 reactions at nitrogen 125 undergoes concerted N to C migration of the alkoxy group leading to formation of the ester and aminonitrene (Scheme 2-15).
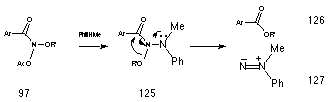
This process is driven by the strong anomeric effect of the high energy nitrogen lone pair in 125 A similar example of this type of rearrangement is found in the thermal decomposition of N,N'-dialkoxy-N,N'-diacylhydrazines 128 which also give esters (Scheme 2-16).
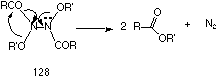
Evidence for consecutive HERON processes in such compounds is presented in Section 2.2.11.1
Thus by analogy with the above reactions, butyl benzoate 118 could form from the N-butoxybenzohydroxamic acid intermediate 108 through alkoxy migration together with simultaneous release of hydroxynitrene 119 in a non-catalysed process. To test for intramolecular formation of ester, a crossover experiment involving two different alkyl N-acetoxybenzohydroxamates was performed. To ensure optimum ester conversion a low acid concentration was used to initiate the reaction at 298K.
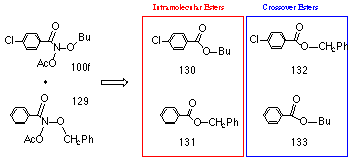
In the joint solvolysis of equimolar quantities of benzyl N-acetoxybenzohydroxamate 129 and butyl N-acetoxy-p-chlorobenzohydroxamate 100f, neither of the mixed, crossover esters 132 and 133 were detected (by 1H NMR) but significant quantities of butyl p-chlorobenzoate 130 and benzyl benzoate 131 were detected. The results were confirmed by HPLC analysis (Table 2-5).
|
Product |
Percentage Yield |
|
benzyl benzoate |
54 |
|
benzyl p-chlorobenzoate |
0 |
|
butyl benzoate |
0 |
|
butyl p-chlorobenzoate |
36 |
Determined by analytical HPLC.
If the observed esters had formed from acid-catalysed esterification from the carboxylic acid and alkanol, butyl benzoate 133 and benzyl p-chlorobenzoate 132 should have been formed. The total lack of these crossover esters indicates that while crossover esterification is possible from the solvolysis products available in the solution, the rate of esterification is very slow under these reaction conditions. Clearly the esters 118 form intramolecularly from the hydroxylated nitrenium ion 108.
Preliminary semi-empirical AM1 calculations by the author and subsequently by Buccigross and Glover177 indicated that such migrations (Scheme 2-14) are concerted and lead to simultaneous formation of ester 118 and hydroxynitrene 119. The activation energies for such migrations are however considerably higher than those involving a nitrogen lone pair; the anomeric driving force of an oxygen lone pair, which is more tightly bound, is considerably weaker. In evidence, Glover found that N,N-dimethoxy-p-toluamide 134 decomposes thermally into alkoxyamidyl 135 and methoxyl 136 radicals at temperatures greater than 110 oC.209
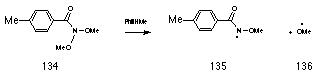
Buccigross found however, that the conjugate anion of the hydroxamic acid 137 rearranges both smoothly and with low activation energy to ester and nitric oxide anion which in acid would give hydroxynitrene.
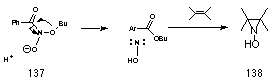
The oxide anion in 137 behaves as a high energy lone pair which results in excellent s*N-O anomeric overlap. Thus it is proposed that the conjugate anion of 108 leads to ester formation in a HERON reaction. This is supported by reactions of butyl N-acetoxybenzohydroxamate 100a in base (See Chapter 3.1) as well as the fact that ester formation drops off dramatically with decreasing pH (Figure 2-16). While hydroxyl migration and formation of butoxynitrene cannot be excluded, the driving force would be low and, like neutral ONO systems, it would be expected to have a high activation energy.
Under neutral or slightly acidic conditions, the by-product of this concerted intramolecular rearrangement would be hydroxynitrene 119. Hydroxynitrenes would be expected to be transient species that dimerise to 1,2-dihydroxydiazene 139.
In acid this would most likely decompose to nitrous oxide 140 and water (Scheme 2-20).194 One method for trapping nitrenes is by addition to double bonds to form aziridines 130 (Scheme 2-19). Several potential problems were envisaged with trapping hydroxynitrene in this manner. These were the low yields expected, and the susceptibility of N-hydroxyaziridines 138 to acid: aziridines, like epoxides, are catalytically opened by acids to form b-amino ethers by alcoholysis,194 a not improbable scenario under these conditions. Nevertheless, attempts were made at trapping hydroxynitrene under conditions conducive to formation of ester in high yield, but neither the aziridine nor products derived therefrom could be isolated or identified. Efforts to identify products derived from hydroxynitrene are continuing in these laboratories.209
HERON reactions - crossover reaction with N-p-chlorobenzoyl-N-butoxy-N'-benzoyl-N'-benzyloxyhydrazine.
As outlined in Section 2.2.11, the HERON reaction is found to occur readily in ONN amides and was first encountered in concerted decomposition of N-anilino-N-alkoxybenzamides formed from SN2 reactions of mutagens with anilines (Scheme 2-15). Subsequent calculations confirmed a low activation energy for this process together with exothermicity brought about by the stability of 1,1-diazenes as well as the esters formed in the process. A report by Cooley et al210 that N,N'-diacyl-N,N'-dialkoxyhydrazines 141 decompose via a concerted four-centre mechanism to ester and nitrogen attracted our attention (Scheme 2-21).
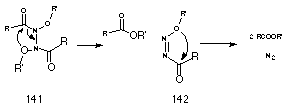
The similarity of the decomposition of these N,N'-diacyl-N,N'-dialkoxyhydrazines 141 to that for N-anilino-N-alkoxybenzamides 125 as well as the proposed decomposition of N-alkoxy benzohydroxamic acid 108 justified a brief review206 of Cooley's work.
N,N'-diacyl-N,N'-dialkoxyhydrazines 141 are formed from the oxidation of hydroxamic esters210 or recombination of N-alkoxy amidyl radicals.211 Cooley postulated that these compounds decompose through two consecutive four-centred process, Scheme 2-21, to give esters and molecular nitrogen with the transition state displaying incipient alkoxide and acylium character. The reaction is favoured by electron-withdrawing substituents on the alkoxy side chain and electron-donating substituents on the acyl-side chain. Since the hydrazine 141 is a special case of a NNO system, the author considered that consecutive three-centred mechanisms would best account for ester formation.
To confirm which pathway was most favourable, an asymmetrical hydrazine, N-p-chlorobenzoyl-N-butoxy-N'-benzoyl-N'-benzyloxyhydrazine 143 was synthesised by lead tetraacetate oxidation of butyl p-chlorobenzohydroxamate 98f and benzyl benzohydroxamate 98a in dichloromethane.212 The asymmetric hydrazine 143 was isolated by flash chromatography from two symmetric products and decomposed thermally in chloroform.
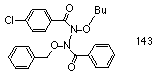
The yields of esters so produced are given in Table 2-7 and clearly indicate that the favoured process involves a three-centred mechanism, the major products being the benzyl benzoate and butyl p-chlorobenzoate esters.
N-p-chlorobenzoyl-N-butoxy-N'-benzoyl-N'-benzyloxyhydrazine 143 was decomposed at 318K in 20% aqueous acetonitrile (D2O-CD3CN) and the progress of the reaction was monitored by 1H NMR through integration of the peaks for the diastereotopic benzyl methylene proton at d5.05.
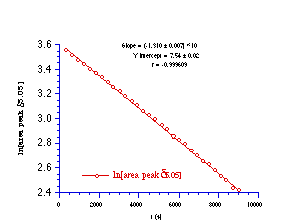
N-p-chlorobenzoyl-N-butoxy-N'-benzoyl-N'-benzyloxyhydrazine 143 decomposed under first order kinetic control as evident from Figure 2-19. The solvolysis reaction was repeated at 298K, 308K, and 331.5K and the results of the Arrhenius study are given in Table 2-6.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The Arrhenius parameters were calculated from a weighted least squares analysis of the data in Table 2-6 (r=0.9990) and the enthalpy of activation was found to be 112.9±3.6 kJmol-1 and the entropy of activation was 26.9±11.6 JK-1 mol-1. This is the first evaluation of the energetics of a HERON reaction process for which a DH of approximately 90 kJmol-1 has recently been calculated using state-of-the-art ab initio methods.213
Interestingly, the small positive entropy change indicates that the transition state is not significantly more disordered than the ground state even though this is a dissociative reaction. This is in accord with a concerted process involving a cyclic transition state in which bond formation and bond breaking is concurrent.
|
Product |
|
|
benzyl benzoate |
|
|
benzyl p-chlorobenzoate |
|
|
butyl benzoate |
|
|
butyl p-chlorobenzoate |
|
Determined by analytical HPLC.
Analysis of the products revealed a similar result to that for the crossover reaction for mixed esters (Section 2.2.11). Clearly the favoured process involves a 3-centred mechanism with the major products being the benzyl benzoate and butyl p-chlorobenzoate esters. However, unlike the mixed ester reaction a small amount of the 4-centred products were detected.
AM1 analysis of 3- centred versus 4- centred pathways
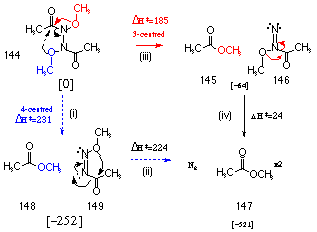
An analysis of the two possible rearrangement pathways for N,N'-methoxy-N,N'-diacetylhydrazine 144 was undertaken at the semi-empirical AM1 level. Scheme 2-22 illustrates the possible decomposition pathways by which N,N'-dimethoxy-N,N'-diacetylhydrazine 144 forms 2 moles of methyl esters 147 and nitrogen gas.
In depth AM1 investigations of the 3- and 4-centred decomposition pathways were carried out as part of this study and later confirmed in a wider investigation of HERON process by Buccigross and Glover177,206
The 4-centred reaction involves a concerted attack from the opposite methoxy moiety across the N-N linkage with concomitant N=N double bond formation (Scheme 2-22(i)). This rearrangement was modelled by decreasing the distance between the alkoxy oxygen and the carbonyl carbon and this ultimately lead to the formation of a stable 1,2-diazene intermediate (Scheme 2-22(i)). An AM1 reaction profile on this molecule was set up such that the energy of the system was determined as the second alkoxy oxygen was set closer to the acyl carbonyl. This did not provide a smooth reaction path but rather the energy rose to a very high level until the molecule snapped to give the products as outlined in Scheme 2-22(ii). This indicated that while the first intramolecular rearrangement was possible, the second step would be unlikely at room temperature and pressure.
A reaction profile analysis on the 3-centred reaction mechanism was conducted using a similar methodology. The bond distance between the alkoxy oxygen and the acyl carbonyl carbon on the same end of the N-N bond. This resulted in a smooth reaction profile to give the 1,1-diazene (Scheme 2-22iii) which in turn decomposed to ester and N2 (Scheme 2-22iv). The latter process required very little activation energy. Semi-empirical methods indicated that the 3-centred, two step process was more likely and required less energy than the single step, 4-centred reaction.
This novel rearrangement is similar to the N to C migrations in N,N-bisheteroatom-substituted amides which undergo the "HERON" rearrangement. N,N-bisheteroatom-substituted amides R-CO-NXY undergo a 3-centre rearrangement in which an electronegative atom (X) migrates from the nitrogen to the carbonyl carbon producing an acyl derivative (R-CO-X) and a substituted nitrene (N-Y). The reaction in these cases appears to be driven by an anomeric effect involving interaction between the lone pair on Y and the X-Ns* orbital. The energy of the transition states for migration is lowered with electron donor substituents on the amide amino substituent and the rearrangement displays high double bond N-N character as well as a build up of charge on the migrating group.
The experimental and theoretical outcomes of this study have subsequently been corroborated by a concurrent investigation by Barton and co-workers who explored this reaction as an effective means of synthesising hindered esters.214