
Theoretical and experimental studies in these laboratories95,105,169,170 have indicated that alkoxy stabilised nitrenium ions should also exhibit substantial oxonium ion character (Scheme 2-23) and so an investigation into the influence of oxygen substituents on oxonium/nitrenium ion formation was undertaken.
Several series of compounds were considered for the study, two of which were phenyl N-acetoxybenzohydroxamates 148 and benzyl N-acetoxybenzohydroxamates (Scheme 2-24). Both series were considered to have excellent potential to change the electronic environment of the oxonium ion through substitution of a range of electron donating and withdrawing para substituents, and hence influence the rate of formation of the nitrenium ion.
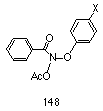
Para-substituted phenyl N-acetoxybenzohydroxamates 148 would allow direct resonance interactions through the ring onto the alkoxy oxygen. The synthesis of aryl benzohydroxamates (95, R=Ar) is non-trivial and only a small number of compounds of this type have being reported. The synthesis of phenyl benzohydroxamate (95, Ar= C6H5, R=C6H5) from potassium benzohydroxamate 94a and diphenyliodonium bromide has been reported in low yield215 and improved by Taylor and Kienzle who reacted thallium(I) benzohydroxamate with diphenyliodonium chloride.216 The synthesis of diaryliodonium salts217,218 is limited in scope and similarly non-trivial.219-222
From these considerations, the synthesis of phenyl N-acetoxybenzohydroxamates 148 was not attempted in the initial phase of the experiments. Benzyl benzohydroxamates 149, on the other hand, could readily be prepared by simple alkylation of potassium benzohydroxamate with benzyl bromides and were found in practise to be easily and conveniently synthesised.
While in these substrates no conduit exists for direct resonance between the para substituents on the ring and the benzyloxy oxygen, inductive effects transmitted through the benzyloxy methylene should be sufficient to influence the electron density on the oxygen and therefore affect the rate of nitrenium ion formation.
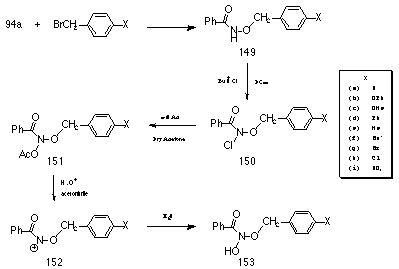
Accordingly a series of para-substituted benzyl N-acetoxybenzohydroxamates 151a-i were synthesised (Scheme 2-24) in an attempt to measure the electronic effect of the alkoxy side chain upon the ease of nitrenium ion 152 formation.
Benzyl benzohydroxamates (149, (c) excepted) were synthesised by the standard method (Scheme 2-1) and were obtained as stable, colourless crystalline materials which could be stored for several months without noticeable degradation.
The substitution reaction involving p-methoxy benzyl bromide or p-methoxy benzyl chloride and potassium benzohydroxamate 94a under a variety of solvent conditions (10% aq. MeOH, 50% aq. MeOH, 100% Dry THF) did not provide p-methoxybenzyl benzohydroxamate 149c. An alternative synthesis was devised that endeavoured to bypass the hydroxamic ester and lead directly to the mutagen p-methoxybenzyl N-acetoxybenzohydroxamate 151c.
O-acetyl-N-benzoylhydroxylamine was synthesised from benzohydroxamic acid and acetic anhydride in dry ether. N-chlorination of the hydroxylamine was achieved by treatment with a 3M excess of t-butyl hypochlorite in dry chloroform and was confirmed by NMR spectroscopy. The sodium salt of p-methoxybenzyl alcohol was obtained by treatment of p-methoxybenzyl alcohol with a 1 molar equivalent of sodium in anhydrous ether under nitrogen and isolated by filtration (under anhydrous conditions). A 1.4 molar excess of the salt in dry acetone was allowed to react with N-acetoxy-N-chlorobenzamide but after stirring at room temperature for 12 hours there was no evidence for the formation of the required product. A third method for the production of the hydroxamic ester, p-methoxybenzyl benzohydroxamate was successfully developed. The nature of the reaction products from the normal alkylation reaction of the potassium salt and benzyl bromide (p-methoxybenzyl methyl ether and p-methoxybenzyl alcohol) indicated that under the reaction conditions resonance stabilised p-methoxybenzyl carbenium ion was formed which was trapped by solvent rather than the potassium salt. Consequently, the reaction conditions were altered to favour SN2 reaction and disfavour the SN1 formation of the benzyl carbenium ion.
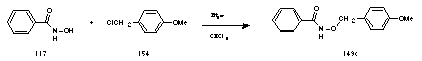
Hence, benzohydroxamic acid 117, p-methoxy benzyl chloride 154 and triethylamine were dissolved and refluxed for 2 hours in dry CHCl3. Workup provided 149c in moderate yield.
N-chlorination of the benzyl benzohydroxamates proceeded smoothly in DCM, with the exception of p-tert-butylbenzyl benzohydroxamate 149f which required 50% DCM/CHCl3 to fully dissolve. Complete N-chlorination was complete within 20-120 minutes.
Acetoxylation of the N-chloro compounds 150a-i was best accomplished using Finkelstein-like conditions194 via Method 2 (Scheme 2-2, NaOAc in acetone) in preference to Method 1 (Scheme 2-2, AgOAc in ether) as side products were reduced leading to increased yields (Table 2-8).
|
Substrate |
|
|
|
151e Me |
|
|
|
151a H |
|
|
|
151g Br |
|
|
|
151h Cl |
|
|
|
151i NO2 |
|
|
a Approximate yield based on 1H NMR of final reaction mixture.
b Quantitative analysis by HPLC.
For instance, treatment of p-nitrobenzyl N-chlorobenzohydroxamate 150i with silver acetate in ether failed to provide the N-acetoxy substrate even after stirring at room temperature for 48 hours, but resulted in the partial formation of p-nitrobenzyl benzohydroxamate 149i. It was thought that the Lewis acid may be insufficiently activating to heterolyse the N-Cl bond in this case and consequently the reaction conditions were adjusted to favour a bimolecular reaction with acetate anion. When p-nitrobenzyl N-chlorobenzohydroxamate 150i was treated with a 1.4 molar excess of anhydrous sodium acetate in dry acetone and stirred at room temperature for 12-48 hours, excellent yields of p-nitrobenzyl N-acetoxybenzohydroxamate 151i were obtained. A molar excess greater than 1.4 appeared to reduce the conversion to N-acetoxy substrates 151. Sodium acetate is only slightly soluble in acetone but Lé Chateliers principle ensures that sufficient soluble acetate anion is available for the SN2 reaction with the N-chloro substrate 150 as the reaction progresses.
Crystallisation of solid sodium chloride above the level of solvent in the reaction vessel flask provided a convenient qualitative indicator that acetoxylation was proceeding smoothly.
Acetoxylation of the benzyl N-chlorobenzohydroxamates 150 resulted in a 0.11 ppm downfield shift for the oxymethylene resonance to about d5.2 which could also be used to monitor the progress of the reaction.
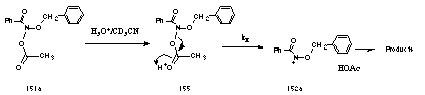
Under acidic conditions in aqueous acetonitrile, benzyl N-acetoxybenzohydroxamate 151a solvolysed following pseudo first-order kinetics giving acetic acid and various solvolysis products (Scheme 2-26). The acid-dependent rate constants of solvolysis at 308K was measured at four acid concentrations and the results are recorded in Table 2-9.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The acid-independent rate constants for solvolysis at 308K was calculated to be (5.59±0.45)x10-3 s-1 (r=0.9936) from the plot of rate constant, k' against hydronium ion concentration.
The rate constant for non-catalysed solvolysis, as given by the intercept, was (1.20±0.86)x10-5 s-1 which is at least two orders of magnitude smaller than the rate constant for acid-catalysis.
The Arrhenius parameters were calculated for the acid-catalysed solvolysis of a series of para-substituted benzyl N-acetoxybenzohydroxamates 151a-i over the temperature range, 298K-338K and the results are given in Table 2-10. k' was obtained at a single [H3O+] for each temperature and kH calculated according to the equation 2-5.
|
Subst. |
|
|
|
|
|
|
r |
|
151c MeO |
44.5(2.6) |
116.7(21.7) |
14286(825) |
118.8(6.9) |
14.87 |
|
|
|
151b PhO |
38.19(2.8) |
64.3(23.3) |
12827(872) |
106.6(7.2) |
3.16 |
|
|
|
151e Me |
47.06(0.71) |
138.0(5.9) |
15690(223) |
130.4(1.9) |
2.062 |
|
|
|
151f But |
43.82(0.94) |
111.1(7.8) |
14731(298) |
122.5(2.5) |
1.821 |
|
|
|
151d Ph |
42.99(0.78) |
104.2(6.5) |
14700(247) |
122.2(2.1) |
0.876 |
|
|
|
151a H |
45.01(1.32) |
121.0(11.0) |
15400(421) |
128.0(3.5) |
0.499 |
|
|
|
151g Br |
40.01(0.45) |
79.4(3.7) |
14181(144) |
117.9(1.2) |
0.239 |
|
|
|
151h Cl |
41.89(1.26) |
95.0(10.5) |
14731(397) |
122.5(3.3) |
0.265 |
|
|
|
151i NO2 |
34.43(1.09) |
33.0(9.1) |
12903(350) |
107.3(2.9) |
0.0573 |
|
|
All compounds solvolysed with a significant activation energy and a positive entropy of activation. Increasing electron donating ability of the para substituent increased the overall rate of solvolysis. The least reactive compound, p-nitrobenzyl N-acetoxybenzohydroxamate 151i, solvolysed with only a small increase in the disorder of the transition state, and with the smallest activation energy. Increasing electron donating ability of the para substituent increased both the energy of activation and entropy of activation. Clearly, the rise in DS is a significant driving force for the reaction, as was the case with the acid-catalysed solvolysis of the butyl N-acetoxybenzohydroxamate 100 series of compounds.
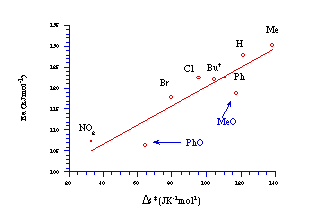
An excellent correlation was obtained when the activation energy of the compounds from p-nitro to p-methyl was plotted against the entropy of activation (r=0.988), however two substrates p-methoxy and p-phenoxy deviated significantly from the expected trend (Figure 2-21). These substituents, with powerful electron donating ability, underwent the fastest solvolysis even though the disorder in the system is lower than expected. As solvolysis to a nitrenium ion is an entropy driven process, a more ordered transition state should result in a slower reaction however this was not observed.
Figure 2-21 indicated that an alternative solvolysis mechanism may be operating when powerful electron donating substituents were present on the benzyl ring. The possibility of an alternative mechanism of solvolysis was supported by an unusual Hammett relationship. When the log of relative rates at 308K (Table 2-10) was plotted, the data revealed an excellent, and unexpected, s+ correlation (r=-1.56; r=0.9797). A s+ relationship normally indicates that para substituents have direct inductive and resonance access, through the molecular framework, to a developing positive charge and the magnitude of the slope of the Hammett plot, r, reflects the ability of the para substituents to influence that charge.
In this system, rates of nitrenium ion-oxonium ion formation should not result in s+ correlation as the influence of mesomeric para substituents must be radically reduced by the intervening benzyloxy methylene group.
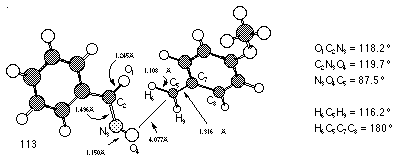
Gas-phase AM1 optimisation of para-methoxybenzyloxynitrenium ion 152c (DH=719 kJmol-1) resulted in fragmentation to a complex of para-methoxy benzyl carbocation (DH=929 kJmol-1) and nitrosocarbonylbenzene (DH=27 kJmol-1) (Figure 2-22). A similar result was obtained using COSMO which simulates a solvated ion. Interestingly, AM1 anticipated a similar decomposition for nitrenium ions 152a (DH=1805 kJmol-1) and 152i (DH=1959 kJmol-1).
Thus preliminary theoretical analysis indicated that benzyloxy stabilised nitrenium ions may fragment to benzyl cation easily suggesting that, in the case of p-methoxy 151c and p-phenoxy 151b substrates, the initial step may involve direct fragmentation to p-methoxybenzyl cation and p-phenoxybenzyl cation. Evidence for such a mechanistic switch was evident from an analysis of the solvolysis products.
The products from the acid-catalysed solvolysis of 151a-e and 151h-i are given in Table 2-11. Since acid strengths were different in each case direct comparisons cannot be made. The range of products from the solvolysis of 151a, 151e and 151h was however similar to those obtained from the butyl N-acetoxybenzohydroxamate with the exception that small quantities of benzoic anhydride were also detected. This is presumably a consequence of reaction of the nitrosocarbonylbenzene 113 intermediate with the benzoic acid 114 it generates with water. Esters are formed in modest but not dissimilar yields from each of 151a, 151e, 151d, 151h and 151i.
|
|
| ||||||
|
|
|
|
|
|
|
|
|
|
Benzaldehyde |
|
|
|
|
|
|
|
|
Benzohydroxamic acid |
|
|
|
|
|
|
|
|
Benzyl benzoate |
|
|
|
|
|
|
|
|
Benzyl alcohol |
|
|
|
|
|
|
|
|
Benzoic acid |
|
|
|
|
|
|
|
|
Benzoic anhydride |
|
|
|
|
|
|
|
At the completion of solvolysis of the p-methoxy substrate 151c, the 1H NMR spectrum was significantly less complex when compared to all other substrates and was dominated by 3 major compounds: benzoic acid; benzyl alcohol; and benzoic anhydride. Benzaldehyde, benzohydroxamic acid and benzyl benzoate were not detected.
Of particular note was the total lack of ester which implied the absence of the N-(p-methoxybenzyl) benzohydroxamic acid 153c; a fundamental intermediate in the proposed mechanism for the solvolysis of all alkyl N-acetoxybenzohydroxamates to alkoxynitrenium ions. For phenoxy substrate 153b the yield of ester was also lower than normal, however both substrates produced significant quantities of benzoic acid anhydride.
A discussion of the results from Table 2-11 has been broken up in the following sub-sections.
From Table 2-11 the yield of benzaldehyde appears to increase with increasing electronegativity of the para substituent from a negligible yield recorded for p-methoxy to a low to moderate yield recorded for p-nitro. From Scheme 2-12, aldehyde is proposed to be formed together with hydroxamic acid from the rearrangement of the protonated alkoxy benzohydroxamic acid intermediate.
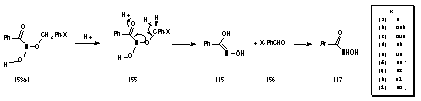
As described previously, this is an acid-catalysed process and the critical point for rearrangement is likely to be protonation of the carbonyl to give 155 and loss of a single benzylic proton to the solution as the molecule reorganises to form the aldehyde and the oxime (Scheme 2-27). As the yield of these products is relatively small, it appears that the para substituents have little of influence over this rearrangement due to their remoteness from the site of protonation. From the yields (Table 2-11) this reaction pathway is clearly overshadowed by other reaction processes.
For the benzyloxy series 151a-i, benzoic acid was formed in moderate to good amounts but the yields reveal no definitive trend (Table 2-11). On the other hand, the yield of benzyl alcohol tends to increase consistently with increasing electron donor capacity of the para substituent.
The p-methoxy substrate 151c gave only 3 major products: benzoic acid; benzyl alcohol and benzoic anhydride. While the formation of nitrosocarbonylbenzene 113 was indicated by the formation of these products, the absence of ester, benzaldehyde and benzohydroxamic acid suggests that N-(p-methoxybenzyl) benzohydroxamic acid 153c is not an intermediate in the reaction.
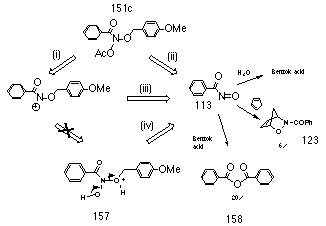
By analogy with the acid-catalysed solvolysis of butyl N-acetoxybenzohydroxamate 100a (Section 2.2.7) N-(p-methoxybenzyl) benzohydroxamic acid 153c should rearrange to p-methoxybenzyl benzoate at low acid concentrations or to benzaldehyde and benzohydroxamic acid at higher acid concentrations. The lack of these products indicate that the hydroxamic ester is not produced and this in turn indicates that formation of nitrenium ion intermediates in the reaction was questionable. The formation of benzoic anhydride 158 in significant yield suggests that nitrosocarbonylbenzene 113 is generated in substantial quantities, either by fast fragmentation of the nitrenium ion intermediate prior to attack by water solvent (Scheme 2-28(iii)), or through direct elimination from the N-acetoxy starting material (Scheme 2-28(ii)).
The presence of nitrosocarbonylbenzene as a solvolysis product was
once again confirmed by trapping the intermediate 113 with
cyclopentadiene as the Diels-Alder cycloadduct,
3-benzoyl-2,3-oxazabicyclo[2.2.1]hept-5-ene 123.
Figure 2-23 a) 3-benzoyl-2,3-oxazabicyclo[2.2.1]hept-5-ene 123 b) Solvolysis of 151c with cyclopentadiene in 25% aqueous acetonitrile.
Cyclopentadiene was dissolved in a solution of 151c, in 25% aqueous acetonitrile, and an acid-catalysed solvolysis reaction was initiated with sulphuric acid at 308K. NMR analysis of the products at completion of the reaction indicated that a similar quantity of benzyl alcohol was produced to the amount that was generated in the absence of the diene. The Diels-Alder cycloadduct, 3-benzoyl-2,3-oxazabicyclo[2.2.1]hept-5-ene 123 was also identified in the mixture by 1H (Figure 2-23b), 13C NMR and was detected in 6% yield by analytical HPLC.
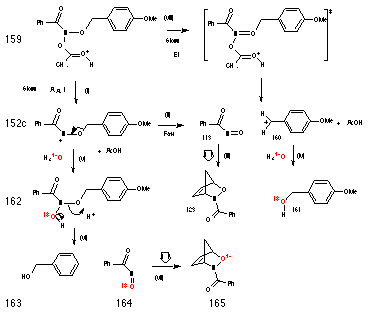
Scheme 2-29 illustrates alternative mechanisms for the decomposition of 159 to give 3-benzoyl-2,3-oxazabicyclo[2.2.1]hept-5-ene 123. The rate determining step could involve concerted release of acetic acid, p-methoxybenzyl carbocation 160 and nitrosocarbonylbenzene 113 in an acid-catalysed E1 process (pathway (viii)). Alternatively, rate determining AAl1 formation of a nitrenium ion 152 could be followed by a fast, highly competitive elimination of p-methoxybenzyl carbocation 160 (pathway (i-ii)). Neither mechanism invalidates the kinetics of the reaction as both processes produce a positively charged intermediate in a pseudo first-order fashion.
Since the nitrosocarbonylbenzene 113 produced by either mechanism would contain the oxygen originating from the benzyloxy substituent, the Diels-Alder adduct that would be formed in the presence of 18O enriched aqueous acetonitrile ought not to display an enhanced M++2 molecular ion in its mass spectrum (Scheme 2-29(iii)). On the other hand, benzyl alcohol 161 that would be formed by trapping of the benzyl cation should be enriched with 18O (Scheme 2-29 (iv)).
Pathway v-vii, involves hydrolysis of the nitrenium ion 152 and based on the study of solvolysis products which revealed the absence of ester, is unlikely to occur. Like solvolysis of butyl N-acetoxybenzohydroxamate 100a, normal acid-catalysed hemi-acetal like decomposition of the hydroxamic acid intermediate 162, Scheme 2-29(vi), would lead to unlabelled benzyl alcohol 163 and labelled nitroso intermediate 164 as well as 18O labelled Diels-Alder adduct 165, Scheme 2-29(vii) in 18O enriched water.
Accordingly, the solvolysis of 151c was repeated in 10% 18O-enriched aqueous acetonitrile (1:4) in the presence of cyclopentadiene. N-benzoyl-2,3-oxazabicyclo[2.2.1]hept-5-ene 123 and benzyl alcohol were then isolated by preparative HPLC. The (M+2)+:M+ ratio for the adduct was less than 0.01 (based on an M+ of 5.2%). The (M+2)+:M+ ratio for the benzyl alcohol of 0.103 was similar to that obtained for labelled benzyl alcohol obtained from the hydrolysis of p-methoxybenzyl bromide in 10% 18O-enriched water (0.122) and an order greater than that for unlabelled p-methoxybenzyl alcohol (0.007). Clearly nitrosocarbonylbenzene 113 is formed by either pathway (i) then (ii) or pathway (viii) but not via acid-catalysed decomposition of the hydroxamic acid 162 (Scheme 2-29(vi)).
The distinction between these pathways was made on the basis of deuterium isotope effects. Unlike pathway (i), pathway (viii) leads to a transition state in which there is a change in hybridisation from sp3 to sp2 at the benzylic carbon. Accordingly a secondary deuterium isotope effect of up to ca. 15% per deuterium is expected.181
Replacement of one or more atoms with their heavier isotopes can bring about a change in the rate in a reacting system and is known as the kinetic isotope effect. Isotopic substitution does not affect the potential energy surface of the molecule, but only influences those properties which are dependant on the atomic masses, such as vibrational frequencies. The extent to which the reaction rate is affected by isotopic substitution is greatest for those of hydrogen H D and T, and has been measured for other isotopes including carbon223-228 12C, 13C and 14C. Since the maximum isotopic rate ratio is approximately the square root of the inverse ratio of isotopic masses, the isotopic effect for atoms heavier than hydrogen becomes very small. A secondary isotope kinetic effect (SKIE) is observed when the rate of the reaction is changed when an atom is replaced by its isotope at a bond which is not broken during a reaction. In a solvolytic reaction, a carbon undergoing a change from sp3 to sp2 hybridisation will benefit from a reduction in the vibrational energy of the bending mode and will favour the reaction as well as favouring hydrogen over deuterium.
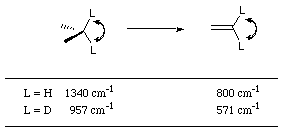
For hydrogen, the reduction in the C-H bending frequency for a
carbon undergoing sp3 to sp2 hybridisation (Figure 2-24) is 540 cm-1 compared to deuterium which is 386 cm-1, which gives ![]() to be approximately 1.4.
to be approximately 1.4.
Acid independent rate constants for acid-catalysed solvolysis of 151c, 151e, 151d and 151i and their bis-deuterated analogues 151j, 151k, 151l and 151m were obtained from the rates of solvolysis at different sulfuric acid concentrations (Scheme 2-30).

Rate determining acid-catalysed elimination of p-methoxybenzyl carbocation 170 involves a change in the hybridisation of the benzyloxy methylene moiety from sp3 in the starting material to sp2 hybridisation in the carbocation (Scheme 2-30(ii)). On the other hand, AAl1 solvolysis to the nitrenium ion 169 does not involve a change in hybridisation for this methylene (Scheme 2-30(i)).
The rate of acid-catalysed solvolysis for the protio and deuterio species are recorded in Table 2-12 below.229
Table 2-12 Rate constants and secondary kinetic isotope effects for para-substituted benzyl N-acetoxybenzohydroxamates at 308K
|
|
|
|
|
|
|
|
|
0.0624 (0.0017) |
|
0.0609 (0.0028) |
|
|
|
|
2.2917 (0.0033) |
|
2.3228 (0.0097) |
|
|
|
|
0.8662 (0.0044) |
|
0.7346 (0.0154) |
|
|
|
|
14.96 (0.54) |
|
11.35 (0.50) |
|
|
*Me, kH from 151e and kD from 151k; ¶Ph, kH from 151d and kD from 151l; NO2,kH from 151i and kD from 151m; # MeO, kH from 151c and kD from 151j;
p-Nitrobenzyl N-acetoxybenzohydroxamate did not show any significant difference in the rate constants for solvolysis (kH/kD=1.02±0.06). Clearly, the rate determining step is well removed from the influence of this carbon, indicating that nitrenium ion formation proceeds via the normal AAl1 mechanism. Interestingly, a similar result was observed when a,a-dideuterio-p-methylbenzyl N-acetoxybenzohydroxamate was solvolysed (kH/kD=0.99±0.01). While elimination of p-nitrobenzyl carbocation from p-nitrobenzyl N-acetoxybenzohydroxamate would be considered unlikely, the positively inductive methyl moiety should increase the possibility for carbocation formation, however this was clearly not the case as the rate constant for the reaction remained the same for both protio and deuterio substrates.
While a para phenyl substituent is more +M but less +I than a para-methyl substituent, the overall effect is to be slightly less electron donating than the methyl moiety. However, the SKIE was determined to be 1.18±0.03, which is consistent with the partial heterolysis of the C-O bond in the rate determining elimination step. Importantly elimination was confirmed for solvolysis of p-methoxybenzyl N-acetoxybenzohydroxamate as the ratio of the slopes gives a kH/kD, of 1.32±0.08, which is close to the theoretical maximum of 1.4181 and is indicative of a transition state with substantial sp2 character at the benzylic carbon (Scheme 2-30(ii)).
The change in mechanism found for p-methoxybenzyl N-acetoxybenzohydroxamate is undoubtedly a consequence of resonance stabilisation imparted to the benzyl cation by the p-methoxy substituent. In contrast, a p-nitro substituent would be expected to disfavour a concerted decomposition and favour reaction by the normal AAl1 mechanism (Scheme 2-30(i)).
It was envisaged that a change in mechanism from AAl1 to E1 would occur with increasing electron donor capacity of the para substituent. A comparison of secondary deuterium isotope effects for some other substrates in the series indicates that this is the case. However there is a clear switch in mechanism in varying the para substituent from methyl (kH/kD = 0.99 ± 0.02) to p-phenyl (kH/kD = 1.18 ± 0.07). Thus the switch from the AAl1 to the E1 mechanism is driven not only by electron donor capacity of the substituents but also by mesomeric stabilisation of the benzyl cation.
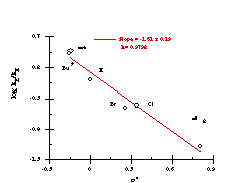
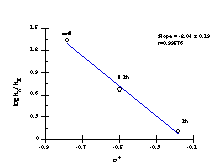
The Hammett data for the benzyloxy series is thus best represented by two independent correlations; one for substrates 151a, 151e-i (Figure 2-25) which correlate with Hammett s substituent constants (r= -1.61 ± 0.19) and one for substrates 151b, 151c and 151d (Figure 2-26) which correlate with s+ (r= -2.04 ± 0.19). The modest sensitivity in the first case is in accord with the inductive interactions with the developing nitrenium/oxonium character at nitrogen and oxygen in the transition state. In the s+ correlation, the sensitivity is in accord with a transition state in which there is a significantly developed, positive charge at the benzylic position. (Scheme 2-30, E1 pathway)
The entire series of substrates have been shown to be significantly mutagenic without metabolic activation (Section 4.3). These are thus direct acting mutagens. Recent results of DNA damage studies230 have indicated that the mutagens modify DNA at N-7 of guanine, the most nucleophilic centre of all the nucleotides. The results in this section have important implications for the mechanism of mutagenesis. Either alkyl N-acyloxy amides behave as molecular electrophiles towards DNA which nucleophilically displaces the acyloxy substituent, or the mutagens generate acylalkoxynitrenium ions. There is a precedent for SN2 displacement of acyloxy group in the work of Campbell and Glover208 who investigated the bimolecular reactions of mutagenic N-acetoxy-N-alkoxybenzamides and N-methylaniline. The SN1 process is however extremely slow and requires an acid catalyst to generate electrophilic nitrenium ions. Under such conditions however, the mutagens 151b, 151c and 151d are likely to yield benzyl cations directly. Thus if catalysed SN1 processes are involved, mutagenicity would have to arise by either alkoxyamidation of nucleotides by nitrenium ions or alkylation of nucleotides by benzyl cations.