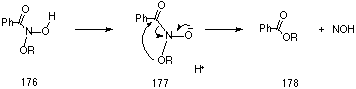
|
|
|
As outlined in Section 2.2.11, in organic medium alkyl N-acetoxybenzohydroxamates 97 have been found to undergo substitution reactions with organic amines.208 The reactions are second-order and lead to unstable N-aminohydroxamic esters which undergo a concerted HERON reaction to give esters and 1,1-diazenes. A similar process has been proposed to explain the formation of esters in the acid-catalysed solvolysis reactions (Section 2.2.11). Solvolysis of alkoxynitrenium ions in these processes leads to N-alkoxyhydroxamic acids 176 which, most probably in the conjugate base form 177, rearrange by an intramolecular process to esters (Scheme 3-1).
176 could also be generated by direct attack of hydroxide ion upon alkyl N-acyloxybenzohydroxamates, and in basic medium would directly generate the conjugate anion. Thus in basic medium, mutagens should form esters. Consequently a study of the behaviour of mutagens in base was conducted.
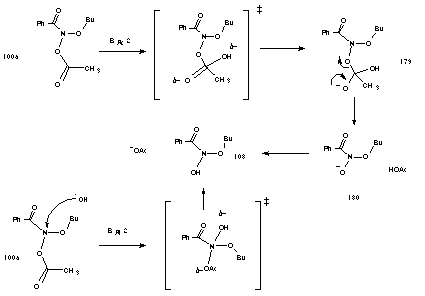
Initially, butyl N-acetoxybenzohydroxamate 100a was treated with an aliquot of concentrated sodium hydroxide solution in 20% aqueous acetonitrile and resulted in the immediate formation of a large amount of butyl benzoate and other minor products. By analogy with the SN2 reactions of Glover and Campbell208 the process most probably involves a fast BAl2 reaction involving hydroxide attack at nitrogen forming labile N-butoxy benzohydroxamic acid 108. Alternatively, hydroxide ions could attack the acetoxy carbonyl in a normal BAc2 process (Scheme 3-2).
The rate equation for a bimolecular reaction between butyl N-acetoxybenzohydroxamate and hydroxide anion is described below.
rate = ![]() = k2[S][OH-] EQUATION
3-1
= k2[S][OH-] EQUATION
3-1
The bimolecular rate constant, k2, could not be determined directly as the disappearance of hydroxide ion with time could not be accurately or conveniently measured over the short reaction period. However by adjusting the reaction conditions the pseudo-unimolecular rate constant k' could be obtained.
Under these conditions, the concentration of hydroxide must be constant throughout the reaction relative to the concentration of butyl N-acetoxybenzohydroxamate 100a or at least sufficiently in excess such that any change in the concentration of hydroxide is insignificant compared to the change in concentration of butyl N-acetoxybenzohydroxamate. In these studies, the ratio of hydroxide to butyl N-acetoxybenzohydroxamate 100a was therefore at least 10:1.
Thus,
Initially the large excess of hydroxide ion necessitated a reduction in the reaction temperature to increase the half-life of the reaction by several orders of magnitude so as to ensure that the disappearance of butyl N-acetoxybenzohydroxamate could be monitored by HPLC. In addition an aqueous acetonitrile mixture was used as solvent.
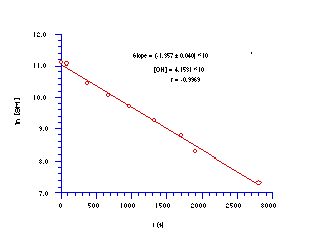
Thus when butyl N-acetoxybenzohydroxamate 100a was solvolysed in 25% aqueous acetonitrile at 275.4K with a 10 molar excess of sodium hydroxide ion the disappearance of butyl N-acetoxybenzohydroxamate was found to be first-order with respect to mutagen concentration (Figure 3-1).
|
104[OH-] (mol.L-1) |
|
|
|
0.10 0.01 |
|
|
1.030.09 |
|
|
1.340.04 |
|
|
2.40.4 |
|
|
2.570.19 |
The reaction was repeated at different hydroxide concentrations under similar pseudo-unimolecular conditions (Table 3-1) and the relationship between k' and [OH-] is illustrated in Figure 3-2.
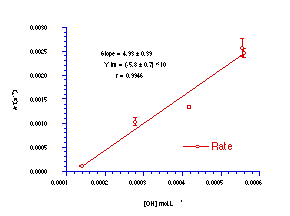
The bimolecular rate constant was obtained from a weighted
least-squares analysis of the data and ![]() was calculated to be 4.93 0.38
Lmol-1s-1. The bimolecular reaction of butyl
N-acetoxybenzohydroxamate with aniline in methanol has been
measured and the Arrhenius parameters have been reported. The
bimolecular rate for this analogous process interpolated to 275.4K
was calculated to be 3 orders of magnitude slower (0.00253
Lmol-1s-1) and is significantly slower, as
would be expected for the weaker nucleophile aniline. While this
reaction proceeds through a bimolecular mechanism, base hydrolysis
could arise through an BAl2 or the normal BAc2
process.
was calculated to be 4.93 0.38
Lmol-1s-1. The bimolecular reaction of butyl
N-acetoxybenzohydroxamate with aniline in methanol has been
measured and the Arrhenius parameters have been reported. The
bimolecular rate for this analogous process interpolated to 275.4K
was calculated to be 3 orders of magnitude slower (0.00253
Lmol-1s-1) and is significantly slower, as
would be expected for the weaker nucleophile aniline. While this
reaction proceeds through a bimolecular mechanism, base hydrolysis
could arise through an BAl2 or the normal BAc2
process.
Butyl benzoate was formed in 80% yield from the base hydrolysis of butyl N-acetoxybenzohydroxamate 100a at 275.5k and the reaction process may be the same as that proposed for the rearrangement of N-butoxy benzohydroxamic 108 under acid-catalysis (See Section 2.2.11). The intramolecular nature of ester formation in that instance was confirmed by joint base hydrolysis of butyl N-acetoxy-p-chlorobenzohydroxamate and benzyl N-acetoxybenzohydroxamate in a crossover experiment (Scheme 2-17).
Butyl N-acetoxy-p-chlorobenzohydroxamate 100f and benzyl N-acetoxybenzohydroxamate 129 were dissolved in 25% aqueous acetonitrile at 298K and treated with an aliquot of dilute aqueous NaOH. Analytical HPLC analysis revealed that the reaction was complete within 60 seconds and confirmed the presence of benzyl benzoate 131 (43.3%), butyl p-chlorobenzoate 130 (46.3%) along with the complete lack of the crossover esters, benzyl p-chlorobenzoate 132 and butyl benzoate 133. This was accomplished by a comparison of retention times and by spiking of the reaction mixture with authentic samples of the esters. These esters are therefore each formed directly from the corresponding parent hydroxamic ester.
N-alkoxy benzohydroxamic acid 176 falls into the class of anomeric amides - amides bearing two heteroatoms at nitrogen.176
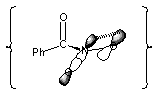
The closely related di-alkoxyamides are predicted by high level ab initio calculations to assume conformations in the ground state in which one N-O s* orbital is co-linear with the 2pz lone pair on the other alkoxy oxygen (Figure 3-3). Such an anomeric interaction weakens one N-O bond.176 AM1 calculations predicts a similar interaction as well as the fact that the concerted HERON migration in these systems is possible but requires a high Ea (215 kJmol-1 in the gas phase).177,206 Thermolysis of N,N-dialkoxy-p-toluamide 134 at temperatures between 100-160oC yield methyl-p-toluate however the ester formation in this case arises not from a HERON reaction but from homolysis to give methoxyl radical and stabilised alkoxyamidyl radicals (Scheme 2-15) which dimerise to hydrazine. As described in Section 2.2.11 such hydrazines readily decompose by the HERON process giving esters.206 Thus the formation of ester by the HERON reaction of the hydroxamic acid is unlikely. The AM1 barrier for such a process is relatively high.177
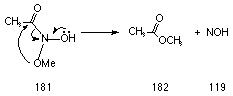
The DH for rearrangement of methoxy in N-methoxy acetohydroxamic acid 181 (Scheme 3-3) is 273 kJmol-1. This is substantially greater than that computed for migration of methoxy in methyl N-dimethylamino acetohydroxamate 183 (Scheme 3-4) which was only 160 kJmol-1 and reflects the difference in energy of an oxygen versus nitrogen lone pair; anomeric effects are weaker where on oxygen lone pair participates.177
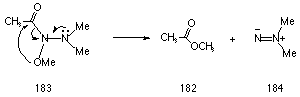
Recent high level ab initio calculations predict a barrier of only 90 kJmol-1 for the corresponding HERON reaction in methyl N,N-dimethylaminoformamide.213
On the other hand, in the conjugate anion form 185, the high energy oxide lone pair interacts strongly with the N-OR s* orbital, thereby driving the rearrangement process which is predicted177 to proceed with low EA (Scheme 3-5).
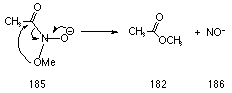
In base, N-butoxybenzohydroxamic acid 108 would be in the conjugate form 180 and facile rearrangement to ester would be expected. The fate of NO- in these reactions has not been determined.
The intermediacy of the conjugate anion of N-butoxy benzohydroxamic acid 180 is further supported by the virtual absence of benzoic acid in the reaction mixtures. HERON rearrangement of hydroxide ion (DH=237 kJmol-1) is predicted by AM1 calculations to be competitive with alkoxy group migration (DH=273 kJmol-1).177 In addition, butoxy nitrene that would be formed by hydroxyl migration could not be detected (vide infra).
Alkaline hydrolysis of butyl N-acetoxybenzohydroxamate 100a at the benzoyl carbonyl (Scheme 3-2) was also precluded by the absence of benzoic acid. Interestingly, small quantities of butanal and butanol were present but benzohydroxamic acid which is formed in concert with butanal under acid-catalysis, was not detected by NMR analysis. Butanal and butanol can form from butoxynitrene. Alkoxynitrenes are very reactive intermediates that have not been characterised spectroscopically but their presence has been confirmed in the oxidation of alkoxyamine through trapping experiments with alkenes at low temperatures.231-238 They have also been prepared and trapped from di-alkoxy amines.239 The oxidation of butoxyamine by lead tetraacetate in the presence of the nitrene trapping agent, 2,3-dimethylbut-2-ene resulted in a 40% yield of N-butoxy-2,2,3,3-tetramethylaziridine.234,240
The absence of any significant amount of butoxynitrene from the base hydrolysis of butyl N-acetoxybenzohydroxamate was confirmed by a trapping experiment. Butyl N-acetoxybenzohydroxamate 100a and 2,3-dimethylbut-2-ene were dissolved in deuterio-acetonitrile (400 mL) and shaken for 20 minutes with sodium hydroxide (100 mL of 2.87 mol.L-1 in D2O). NMR analysis of the reaction mixture at completion indicated complete loss of starting material together with a good yield of butyl benzoate (78%). Small amounts of butanol and butanal were present, but absence of N-butoxy-2,2,3,3-tetramethylaziridine was confirmed by comparison with the NMR resonances of authentic material, prepared via the standard procedure.234 The source of butanal and butanol was not determined.
Clearly, the absence of benzoic acid along with the failure to trap butoxynitrene as N-butoxy-2,2,3,3-tetramethylaziridine indicates that butoxy migration is clearly favoured over hydroxy migration in the rearrangement of N-butoxy benzohydroxamic acid.
The bimolecular reaction of hydroxide ion and butyl N-acetoxybenzohydroxamate 100a has been reported in Section 3.1. The pseudo-unimolecular reaction rate constant was measured at 275.4K and was significantly faster than that found for the bimolecular reaction of 100a with aniline in methanol. The actual position of hydroxide attack could not be unequivocally determined in these experiments but the SN2 reaction at nitrogen was confirmed from a study of the reaction of hydroxide on the benzyl N-benzoyloxybenzohydroxamate 172 series of compounds.
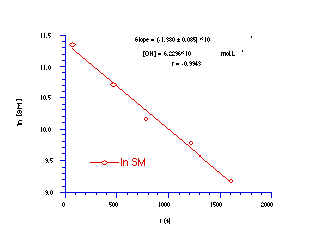
Like butyl N-acetoxybenzohydroxamate 100a, benzyl N-benzoyloxybenzohydroxamate 172a hydrolyses under pseudo first-order kinetic control when treated with a 10 molar excess of hydroxide ion in 25% aqueous acetonitrile at 275.4K as evidenced by the straight line relationship between ln[SM] and time (Figure 3-4).
|
|
|
|
|
1.3800.085 |
|
|
2.4220.119 |
|
|
1.4180.103 |
|
|
0.5080.142 |
The rate of hydrolysis of 151a was measured at four different hydroxide concentrations (Table 3-2) to obtain the pseudo first-order rate constant at 275.4K and the results are plotted in Figure 3-5.
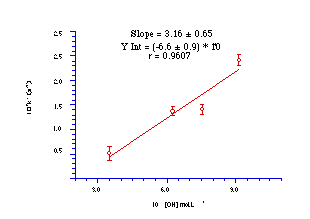
While there is some scatter in the rate constants, a linear dependence upon [-OH] is demonstrated and the rate constant at 275.4K was found from the slope to be 3.160.65 Lmol-1s-1, slightly slower than that for butyl N-acetoxybenzohydroxamate (4.930.39 Lmol-1s-1).
The bimolecular rate constants for the solvolyses of 172a,c-e,g,h at 275.4K are presented in Table 3-3.
|
Substrate |
kx (s-1) |
|
kx/kH |
log(kx/kH) |
|
172c Me |
2.27 (0.39) |
|
0.718 (0.196) |
-0.144 (0.135) |
|
172a H |
3.16 (0.65) |
|
1.000 (0.291) |
0.000 (0.149) |
|
172d Cl |
3.82 (0.69) |
|
1.207 (0.331) |
0.0818 (0.105) |
|
172e CHO |
4.91 (0.59) |
|
1.551 (0.370) |
0.191 (0.118) |
|
172g CN |
6.97 (0.09) |
|
2.204 (0.455) |
0.343 (0.100) |
|
172h NO2 |
8.14 (1.26) |
|
2.571 (0.663) |
0.410 (0.129) |
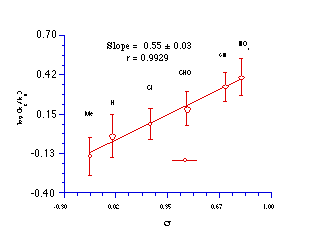
Relative rate constants at 275.4K give an excellent correlation with the Hammett s substituent constants but with a relatively small, positive reaction constant (r=0.55). This indicates that electron-donating substituents decrease the rate of solvolysis, while the moderate magnitude for r reflects only limited influence of substituents on the stability of the transition state. Such lack of sensitivity is characteristic of SN2 processes in which electron donor groups in the leaving moiety (L) inhibit nucleophilic attack but favour heterolysis of the Y-L bond (Scheme 3-6).
The situation is very different for base-catalysed solvolysis of benzoate esters. These reactions are relatively sensitive to the electronic effect of para substituents. The rate determining step is addition of -OH to the carbonyl to give a tetrahedral intermediate (Scheme 3-2), a process that is strongly directed by the positive character of the carbonyl carbon.181 Thus the Hammett reaction constants for the base hydrolysis of alkylbenzoates in aqueous organic solvents are typically 2.1 - 2.3.181
BAl2 reactions, while rare, have been reported for the reactions of ethyl benzenesulphonates with ethoxide241, methyl esters with trimethylamine242 and methyl esters with methoxide.243 Hammett reaction rate constants have not been published for these reactions but by revisiting the data of Morgan's et al from the reaction of ethyl para-substituted benzenesulphonates with ethoxide241 the reaction rate constant for the s relationship of this BAl2 process was calculated to be only 1.4 (r=0.988). The lower reaction rate constant in this study is in accordance with the para substituents exerting a lower influence over the reaction centre. In addition, the Hammett reaction constant is similar to that obtained for the SN2 reaction of N-methylaniline at nitrogen with the same series of benzyl N-benzoyloxybenzohydroxamates. Campbell and Glover found that in methanol, rate constants for the reaction also correlate with Hammett s constants with r = 0.1.244
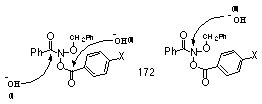
Hydroxide attack at the benzamide carbon (Figure 3-7(i)) would not lead to the hydrolysis products, para-substituted benzoic acid and benzyl benzoate, and can be discounted on these grounds. In addition this process would be insensitive to para substituents on the benzoate group. Thus it can be concluded that base hydrolysis occurs by an SN2-type reaction at nitrogen (BAl2 ester hydrolysis, Figure 3-7(iii)) rather than through conventional attack at the carbonyl (BAc2 ester hydrolysis, Figure 3-7(ii)) and parallels the reactions of alkyl N-acyloxybenzamides with organic nucleophiles. Mutagens should therefore be susceptible to reactions with nucleophilic substance intracellularly and to binding to DNA through nucleophilic centres on nucleotides. Adventitious interception by amines and other basic entities intracellularly can be expected.
Alkyl N-acyloxybenzohydroxamates react with hydroxide in a bimolecular process at nitrogen. The process is fast and follows pseudo first-order kinetics. The major hydrolysis product is the corresponding alkyl benzoate ester formed by HERON reaction involving the conjugate anion of the intermediate hydroxamic acid.